Innate defenses against HIV

HIV-1 capsid pattern sensing by cellular proteins
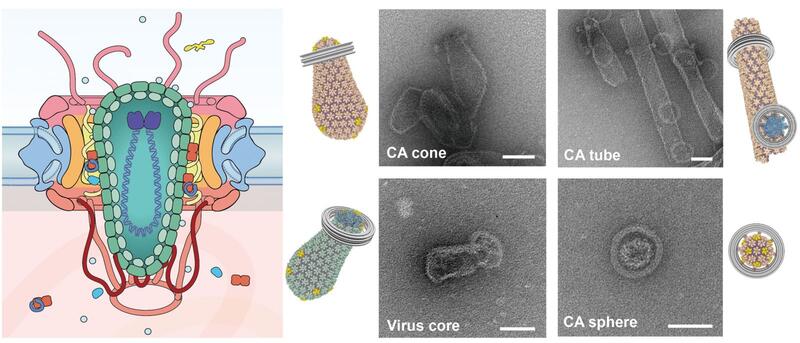
A major focus in the lab is investigating the interactions between the HIV and numerous cellular factors through the course of early HIV infection. The HIV capsid houses the viral genome and consists of over a thousand copies of capsid proteins (CA) arranged into hexamer and pentamer building blocks. These hexamers and pentamers further associate with each other to form a cone-shaped capsid structure. Cellular factors may interact with the capsid core to block infection (restriction factors), whereas others may be recruited by the virus to promote infection (cofactors).

A common theme of these interactions is the host pattern sensing of HIV capsid. Most capsid-binding proteins show relatively little affinity for the monomeric CA. Instead, they recognize the interfaces between multiple CA subunits, or more high-order interfaces between multiple hexamer/pentamer building blocks. However, this recognition mode has greatly limited high-resolution structural characterization of host-capsid interactions because it is difficult to reconstitute the pattern within the cone-shaped structure for uniform binding of host factors in vitro.
To overcome this major challenge, we have used a variety of protein engineering tools to build a library of assembled CA oligomers. These CA oligomers faithfully recreate the native high-order interfaces found between CA subunits and can be purified in large quality. These tools have enabled us to map how important viral restriction factors and co-factors bind the capsid. Among the host factors amenable to our engineered tools are TRIM5α, TRIMCyp, MxB, Fez1, SUN2, and numerous small-molecule compounds. Excitingly, we can use these tools to discover new host factors, and streamline their biochemical characterization. Furthermore, we are pursuing high-resolution structures of CA-host factor complexes using crystallography and cryo-EM.
A first line of defense: TRIM5 proteins
Retroviruses are often limited to a small number of host organisms due to species-specific cellular restriction factors. The tripartite motif proteins TRIM5α and TRIMCyp are important components of the cross-species barrier to HIV and many other retroviruses. TRIM5α inhibits retrovirus infection by directly recognizing retroviral capsid in a species-specific manner and eliciting premature disassembly of the capsid and activation of cellular innate immune signaling. Functioning as a viral capsid pattern sensor, TRIM5α binds only to the assembled capsid lattice. Our goal is to investigate TRIM5 protein-capsid interactions in vitro and establish the structural basis of these interactions.
HIV restriction by MxB
The myxovirus resistance (Mx) proteins are interferon-induced dynamin GTPases that can inhibit a variety of viruses. The well-characterized MxA protein is known for inhibiting influenza viruses. More recently, MxB was shown to restrict HIV-1 by an unknown mechanism that likely occurs in close proximity to the host cell nucleus and involves the viral capsid. We have determined the crystal structure of MxB and revealed determinants involved in HIV-1 restriction. Although MxB is structurally similar to MxA, their antiviral functions rely on separate determinants, indicating distinct mechanisms for virus inhibition. Additionally, MxB directly binds the HIV-1 capsid and this interaction depends on dimerization and the N-terminus of MxB as well as the assembled capsid lattice. These insights establish a framework for our future investigation of the mechanism by which MxB restricts HIV-1.
Mutation of viral DNA by APOBEC3 proteins
APOBEC3G (A3G) is a host cytidine deaminase which extensively mutates viral DNA to restrict HIV infection. To elude this host defense, HIV virion infectivity factor (Vif) binds A3G and targets it for proteosomal degradation. Our goal is to first establish the mechanism by which A3G specifically recognizes viral DNA. At the same time, we’re also investigating the biochemical and structural principles underlying the interaction between Vif and A3G. Information gained from these studies will be used to direct structure-based design of anti-HIV drugs. Screening of Vif inhibitors is being carried out at the Yale Small Molecule Discovery Center.
Suppression of reverse transcription and regulation of cellular dNTPs by SAMHD1
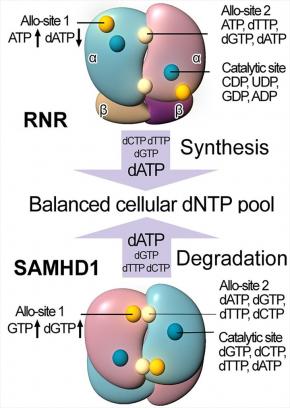
SAMHD1, a deoxyribonucleoside triphosphate triphosphohydrolase (dNTPase), prevents the infection of non-dividing cells by retroviruses,including HIV, by depleting the cellular dNTP pool available for viral reverse transcription. SAMHD1 is a major regulator of cellular dNTP levels in mammalian cells. Mutations in SAMHD1 are associated with chronic lymphocytic leukemia (CLL) and the autoimmune condition Aicardi Goutières syndrome (AGS). We have obtained extensive biochemical and structural data that uncover an exquisite activation mechanism of SAMHD1 via combined action of both GTP and dNTPs at the eight allosteric and four catalytic sites of the SAMHD1 tetramer. These results establish a complete framework for our future studies of the important functions of SAMHD1 in the regulation of cellular dNTP levels, as well as in HIV restriction and the pathogenesis of CLL and AGS.
Viral hijacking of membrane trafficking by Nef
The HIV protein Nef interacts with a diverse range of cellular factors to enhance viral fitness and infectivity. Nef mediates the association of major histocompatibility complex class I (MHC-I) with the clathrin adaptor protein complex 1 (AP-1), leading to the delivery of MHC-I to the endosome and lysosome via clathrin-coated vesicles. Similarly, Nef re-localizes CD4, and the recently discovered SERINC family of proteins, from the plasma membrane via hijacking of AP-2 in order to evade these potent restriction factors. We aim to elucidate the structural and biochemical principles of how HIV Nef co-opts the cellular membrane trafficking machinery to benefit the virus.
The last line of defense: enveloped virus tethering by BST2
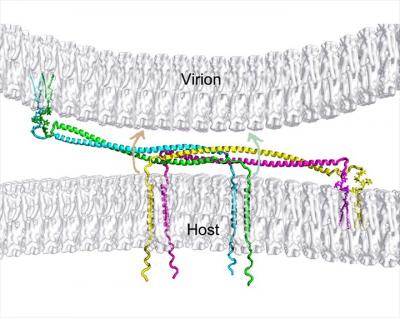
HIV completes its life cycle by budding off the host cells for the next round of infection. The host transmembrane protein BST-2/CD317 (tetherin) tethers HIV, and other enveloped viruses as well, to the cell preventing their release from the cell surface. HIV evades the host defense by the viral protein u, Vpu, which antagonizes tetherin restriction by an unknown mechanism. Our goal is to establish the chemical and structural principles by which tetherin restricts HIV release and the mechanisms by which Vpu antagonizes tetherin. Information gained will be useful for the design of new antiviral therapeutics.
Nuclear entry of HIV capsid
The delivery of HIV-1 genome to the nucleus is an indispensable step in the infection process but the mechanistic insight of this process is scarce. One of the most exciting recent developments in the HIV-1 field is the possibility of the viral capsid staying intact and carrying the HIV-1 genome into the nucleus. Nuclear transport is tightly regulated by the nuclear pore complex (NPC), a massive protein complex of ~120 MDa. However, whether the ~60 nm wide HIV-1 capsid can passage through the NPC pore of ~40 nm in diameter remains unanswered. Our overall goal is to elucidate the mechanistic details of HIV-1 nuclear entry using a cell-free model system that robustly recapitulates the native environment of nuclear pores. To achieve this goal, we have established a powerful experimental platform that combines two cutting-edge techniques: a DNA-origami mimic of the NPC central channel (termed NucleoPorins Organized by DNA, or NuPOD) and programmable HIV-1 capsid assemblies that faithfully recreate the HIV-1 capsid surface.