
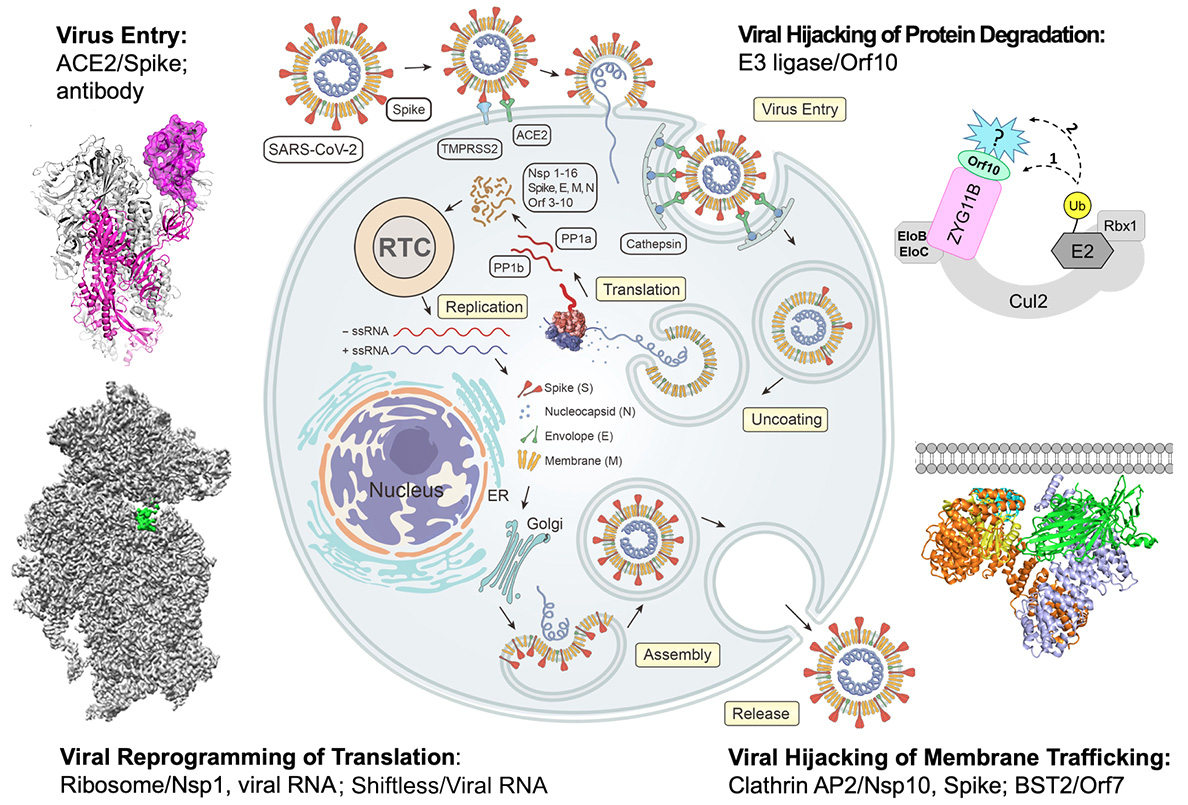
Host interactions with SARS-CoV-2
Blocking virus entry
The SARS-CoV-2 virus gains cell entry through interaction between its spike glycoprotein and the human Angiotensin-converting enzyme 2 (ACE2). We aim to uncover the detailed mechanisms of ACE2-mediated viral entry, understand the processing of the viral spike glycoprotein by the cellular serine protease TMPRSS2, and identify antibodies and small molecules that disrupt the interaction between the spike protein and ACE2/TMPRSS2. We will use biochemical and structural techniques to uncover mechanistic insights of the spike protein interactions with these antibodies and small molecules, which will facilitate the design of vaccines and therapeutics to block the cell entry of coronavirus.
Nsp1-mediated reprogramming of cellular translation toward viral RNA
Nonstructural protein 1 (Nsp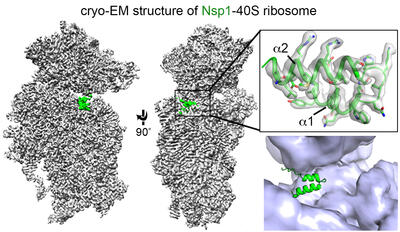 1) of SARS-CoV-2 plays a key role in suppressing host gene expression. We demonstrated that Nsp1 causes the most severe cytopathic effect and significantly alters multiple gene expression programs in human lung cells, physically blocks the mRNA entry channel and locks the 40S ribosomal subunit in a conformation incompatible with mRNA loading. Our goal is to establish mechanistic structural bases for how SARS-CoV-2 ensures translation of its own mRNA while global protein synthesis is shut down by Nsp1, and fully understand the cellular consequences of Nsp1 expression.
1) of SARS-CoV-2 plays a key role in suppressing host gene expression. We demonstrated that Nsp1 causes the most severe cytopathic effect and significantly alters multiple gene expression programs in human lung cells, physically blocks the mRNA entry channel and locks the 40S ribosomal subunit in a conformation incompatible with mRNA loading. Our goal is to establish mechanistic structural bases for how SARS-CoV-2 ensures translation of its own mRNA while global protein synthesis is shut down by Nsp1, and fully understand the cellular consequences of Nsp1 expression.
Inhibiting viral programmed ribosomal frameshifting
Programmed ribosomal frameshifting (PRF) is an indispensable feature of the SARS-CoV-2 life cycle wherein two separate sets of viral proteins are produced from the same viral mRNA. PRF is also a critical step in the life cycles of many other viruses, including HIV-1. A cellular innate immune factor called ‘Shiftless’ was recently identified to be a broad-spectrum inhibitor of programmed ribosomal frameshifting, but the structural and mechanistic details of its function are unknown. To unravel these mechanistic details, we are currently undertaking biochemical and structural studies of Shiftless with SARS-CoV-2/HIV-1 PRF signal RNAs and human 40S/60S/80S ribosomes.
Viral hijacking of host protein degradation pathways
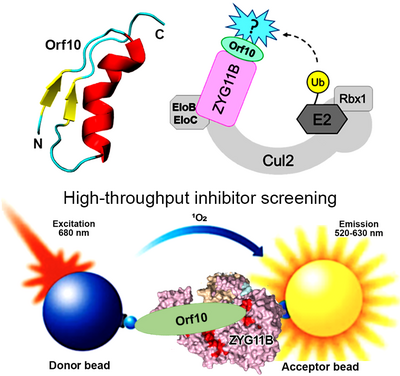
Cellular protein degradation pathways are common targets that viruses hijack to escape host immune restrictions. Recent proteomic analyses have suggested that SARS-CoV-2 Orf10 specifically interacts with the human CRL2ZYG11B complex, a member of the Cullin-RING E3 ubiquitin ligase family that plays a central role in proteasome-mediated degradation of cellular proteins. This interaction likely targets yet unidentified host restriction factors for degradation. We aim to identify the role of SARS-CoV-2 Orf10 during viral infection as well as molecular mechanisms by which it interacts with the human CRL2ZYG11B complex. This information will be harnessed to screen small molecule inhibitors to disrupt the Orf10-CRL2ZYG11B interaction, facilitating the development of novel drugs for COVID-19 therapy.
Viral hijacking of host membrane trafficking pathways
SARS-CoV-2 and other viruses are tasked with subverting or hijacking cellular membrane trafficking pathways to remove membrane associated immune molecules from the site of action. One such host defense protein, BST2, serves to restrict viruses by acting as a molecular tether between the host cell membrane and budding viral membrane; thus, progeny virions are severely impaired in their ability to travel and infect new cells. We aim to understand how SARS-CoV-2 Orf7a antagonizes the effect of BST2 tethering during infection. In addition, we are investigating the roles of Nsp10 and the spike glycoprotein in modulating clathrin-mediated vesicular trafficking pathways to ensure efficient viral replication.